
| Phone: |
+420 54949 4947, +420 54949 2685, +420 54949 2685 |
| E-mail: |
|
| Office: |
|
Research areas
-
Structure and dynamics of proteins, nucleic acids, saccharides, and their complexes in relationship to their biological function
-
Mechanistic studies on enzymatic reactions
-
Chemoinformatics and structural bioinformatics
-
Structure and dynamics of supramolecular complexes
Main objectives
-
To study the therapeutical and bioanalytical aspects of recognition and adhesion phenomena in host-pathogen interactions
-
To develop new methodologies for investigating the structure, interactions, and dynamics of biomolecules
-
New tools for structural bioinformatics and chemoinformatics
-
Development of soft matter models and understanding of self-assembled bimolecular systems
Content of research
Computational chemistry helps to interpret experimental observations or to get a new information about systems where experimental approaches are not applicable or difficult to apply. It is widely employed in, for example, design and studies on new molecules with a possible biological impact.
Structure and Dynamics of Biomolecules
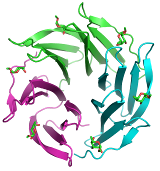 We use a wide spectrum of state-of-the-art computational techniques to study structure and dynamics of short peptides, proteins, enzymes, and short nucleic fragments. Our main attempt is to understand how these biomolecules are assembled in space and how their structure is related to their function. We use a molecular docking to predict unknown structures of complexes between proteins and small molecules. Interactions are studied by several approaches ranging from very precise quantum chemical calculations to methods employing molecular mechanics.
We use a wide spectrum of state-of-the-art computational techniques to study structure and dynamics of short peptides, proteins, enzymes, and short nucleic fragments. Our main attempt is to understand how these biomolecules are assembled in space and how their structure is related to their function. We use a molecular docking to predict unknown structures of complexes between proteins and small molecules. Interactions are studied by several approaches ranging from very precise quantum chemical calculations to methods employing molecular mechanics.
Enzymatic Reactions
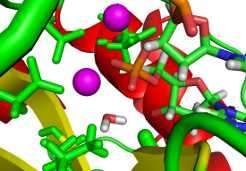 The understanding of reaction mechanisms is essential step in rational design of enzyme inhibitors that might act as drugs. We employ hybrid quantum mechanics (QM) / molecular mechanics (MM) approach to find the most probable reaction pathways. Various techniques to explore complicated potential (free) energy surfaces are used. They range from single/double coordinate energy scans to advanced techniques employing free energy calculations and Car-Parrinello dynamics. Developed techniques are used to study nucleases, glycotransferases and glycohydrolase enzymes.
The understanding of reaction mechanisms is essential step in rational design of enzyme inhibitors that might act as drugs. We employ hybrid quantum mechanics (QM) / molecular mechanics (MM) approach to find the most probable reaction pathways. Various techniques to explore complicated potential (free) energy surfaces are used. They range from single/double coordinate energy scans to advanced techniques employing free energy calculations and Car-Parrinello dynamics. Developed techniques are used to study nucleases, glycotransferases and glycohydrolase enzymes.
Supramolecular Chemistry
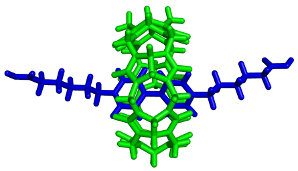 Structure, stability and reactivity of supramolecular systems are studied by molecular modeling techniques, including both quantum and molecular mechanics approaches. Key molecules in our projects are glycoluril oligomers such as cucurbit[n]urils and bambus[n]urils. We study their interactions with various organic and inorganic guests. Our main attention is focused on reliable description of forces leading to complex assembly, which might be used in rational host modifications providing desired properties.
Structure, stability and reactivity of supramolecular systems are studied by molecular modeling techniques, including both quantum and molecular mechanics approaches. Key molecules in our projects are glycoluril oligomers such as cucurbit[n]urils and bambus[n]urils. We study their interactions with various organic and inorganic guests. Our main attention is focused on reliable description of forces leading to complex assembly, which might be used in rational host modifications providing desired properties.
Coarse Grained Simulations
 We study cellular uptake of nanoparticles and virus capsids. These processes are crucial for drug delivery, toxicity, and nanomedicine. We investigate conditions of passive endocytosis of ligand-coated particles of various sizes, shapes, and coverage across a zero-tension, receptor-rich, phospholipid membrane. We also develop new coarse grained models for studying self-assembly of rod like molecules (peptides, carbon nanotubes, etc.). Using these models we determine crucial parameters that lead to different aggregated morphologies including barrels, fibrils, ribbons, bilayers and other oligomers.
We study cellular uptake of nanoparticles and virus capsids. These processes are crucial for drug delivery, toxicity, and nanomedicine. We investigate conditions of passive endocytosis of ligand-coated particles of various sizes, shapes, and coverage across a zero-tension, receptor-rich, phospholipid membrane. We also develop new coarse grained models for studying self-assembly of rod like molecules (peptides, carbon nanotubes, etc.). Using these models we determine crucial parameters that lead to different aggregated morphologies including barrels, fibrils, ribbons, bilayers and other oligomers.
Chemoinformatics and Structural Bioinformatics
 Nowadays, a large amount of information about biomolecules (i.e. sequence of DNA, structure of proteins) and about small molecules (drug-like molecules, ligands, etc.) is available. Main goal of bioinformatics and chemoinformatics research is a processing of these data, which can provide information very useful in pharmacy, medicine, biotechnology etc. Our laboratory is focusing on advanced analyses of protein 3D structures, processing data from next generation sequencing and predicting of physico-chemical properties of organic molecules.
Nowadays, a large amount of information about biomolecules (i.e. sequence of DNA, structure of proteins) and about small molecules (drug-like molecules, ligands, etc.) is available. Main goal of bioinformatics and chemoinformatics research is a processing of these data, which can provide information very useful in pharmacy, medicine, biotechnology etc. Our laboratory is focusing on advanced analyses of protein 3D structures, processing data from next generation sequencing and predicting of physico-chemical properties of organic molecules.
RESEARCH PROJECTS
Computational studies of protein-carbohydrate interactions
-
Prediction of ligand position by molecular docking
-
Molecular dynamics simulations, methods for binding free energy prediction
-
High throughput virtual screening for lectin inhibitor design
-
High level QM methods to quantify the role of dispersion energy and CH-π interactions
-
In silico protein engineering of lectin mutants with improved affinity/selectivity
-
Thermodynamic integration and other methods for binding free energy prediction
QM/MM studies of reaction mechanisms of endonucleases and glycosyltransferases
-
QM/MM methods to study reaction mechanism of glycosyltransferases
-
QM/MM methods to study reaction mechanisms of endonucleases
-
Free energy calculations to identify reaction pathways in enzymatic reactions of endonucleases
-
Reactive force-field molecular dynamics to study reaction mechanism of glycosyltransferases
Supramolecular chemistry of cucurbiturils and bambusurils
-
Free energy calculations based on potential of mean force methods to reproduce results from experimental NMR studies
Coarse grained simulations
-
Assembly of peptides in solution
-
Interaction of peptides with membranes
-
Interaction of nanopartticles with membranes
Cheminformatics and structural bioinformatics
-
Empirical methods for fast calculation of partial atomic charges
-
Development of pKa predicting QSPR models based on atomic charges
-
Finding, comparing and characterization of protein structural motifs
Software development
CURRENT RESEARCH INFRASTRUCTURE
The group operates with a research infrastructure composed of computational clusters with about 1000 processor cores and the possibility of sharing National Academic Supercomputer Centre resources.
1. Engineering the glycans of stem cells to have required bioactivity – a computational study combined with an experiment
Supervisor: prof. RNDr. Jaroslav Koča, DrSc.
Consultants: Ing. Igor Tvaroška, DrSc.
Annotation:
Investigations throughout the end of the 20th century revealed that cell migration is coordinated by chemoattractants present within endothelial beds. The cell migration is encoded by a series of overlapping steps, and tissue-specific migration is controlled by a discrete combination of various receptors present on circulating cells. The first step of the cascade, tethering contact of cells on the endothelium, is governed by interactions of selectins (E-, P-, and L-selectin) with their ligands. Selectin ligands are specialized carbohydrate determinants, consisting of sialofucosylations containing an alpha(2,3)-linked sialic acid substitutions and an alpha(1,3)-linked fucose modification displayed as the tetrasaccharide sialyl Lewis X (sLex) that are exhibited on the glycoform CD44. However, in some human mesenchymal stem cells, the terminal alpha(2,3)-sialyllactosamine moieties are lacking alpha(1,3)-fucosylation at N-acetylglucosamine to have the complete sLex determinant and, therefore, are lacking E-selectin ligand. The enzyme responsible for the alpha(1,3)-fucosylation is fucosyltransferase VI (FucTVI). The Ph.D. study will attempt to shed some light on the role of alpha(1,3)-fucosylation on a function of selectin ligands.
Keywords: chemoattractants, alpha(1,3)-fucosylation, ligands selection, glycans
2. Partial atomic charges – a clue to predict chemical and biological behaviour of biomacromolecules
Supervisor: prof. RNDr. Jaroslav Koča, DrSc..
Annotation:
Nowadays, large amount of structural data about biomacromolecules is available and the number of resolved structures is growing rapidly. This information provides us a great opportunity to analyse the data and to reach a key biological information. Partial atomic charges belong to very promising molecular characteristics. They describe the distribution of electron density in a molecule, and, therefore, they provide clues regarding the chemical and biological behaviour of molecules. Thanks to advanced computational approaches, they can became available also for large and extra-large biomacromolecules. As they belong to biomacromolecular chemical characteristics, they can consequently be very helpful in revealing biological implications. For example, they can serve for a prediction of point mutations effect, for understanding allostery and other biomacromolecular activation mechanisms, sometimes processes that are very difficult to access by experimental approaches.
The thesis will focus on two challenges. First is development a methodology to calculate atomic charges in biomacromolecular systems in a reasonable time and with a reasonable accuracy, and second is application of atomic charges on revealing selected biological problems where such biomacromolecules are involved.
Keywords: biomacromolecules, partial atomic charges, data analysis
3. Characterizing sequence-structure-function relationships: A multi-dimensional approach for evaluating similarity in structural biology data
Supervisor: prof. RNDr. Jaroslav Koča, DrSc.
Consultants: Mgr. Ing. Crina-Maria Ionescu, Ph.D.
Annotation
Understanding the similarity between the sequence and/or structure of biomolecules has been key to inferring evolutionary relationships and predicting the function of biomolecules. We propose a novel approach for defining and evaluating the similarity between biomolecules. The approach is based on representing structural biology data as a square matrix of biomolecular structure elements and their properties; we then define similarity as the result of comparing the eigenvalues and eigenvector spaces determined for the biomolecules compared. Such a test of statistical similarity is intended to determine whether the compared biomolecules share features of structure, function, biological pathway, or affinity for a certain range of binding partners.
Keywords: biomolecules, sequence-structure-function relationship, data analysis












