


Free energy calculations based on potential of mean force methods to reproduce results from experimental NMR studies
The main aim of the project is an efective implemenattion of methods for free energy calculation. Free energy is the one of the most importatnt state quantity for biochemical processes description. Multidimensional reaction coordinate driving of system is one part of a project. Driving means, we are able to influenece system in desired way to explore interesting areas of phase space in sense of possible biochemical processes and state change. Current used software packages (AMBER) are able to run molecular simulations in parallel thus implementation of mentioned methods has to be parallelized as well. One of the entire part of the project is testing and case study on selected biochemical systems. At present pseudorotaxane complexe was selected for a study.
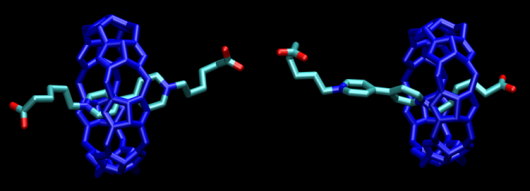
Figure 1: Two possible states of pseudorotaxane molecule. Fully interlocked state, and partially interlocked state have different energy. Energy levels of these two states can be switched when change of environmental pH occurs.
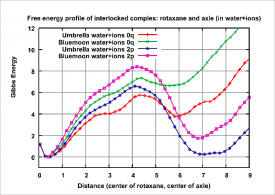
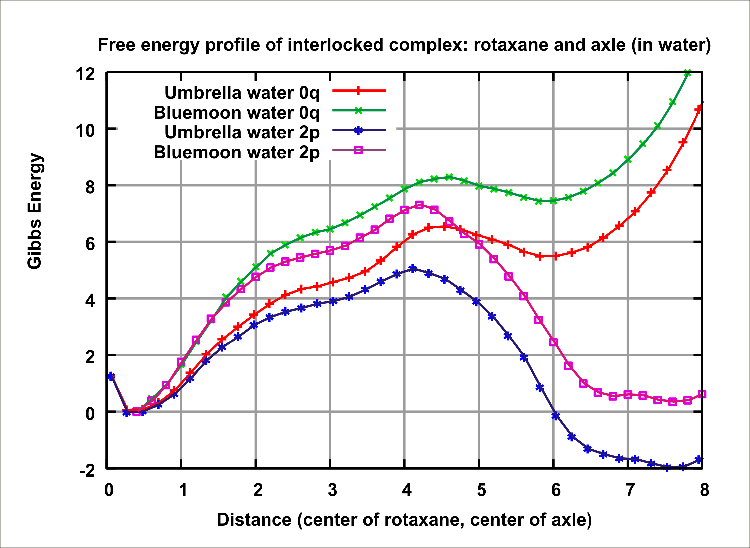
Figure 2: Resultant graph of free energy profile of pseudorotaxane complex. For unprotonated case of system (0q), preferable state is at distance 0A (full interlocation). In protonated case of system (2p), preferable state is at distance 7.5A (partial interlocation).

